Nutrition coalition shared a paper with me recently & I was surprised to find that current nutrition advise for familial hypercholesterolemia (FH) is evidence-free! I extracted the key points from above paper & pasted below:
Atherogenic dyslipidaemia risk triad: triglycerides, high-density lipoprotein (HDL) and small, dense LDL.
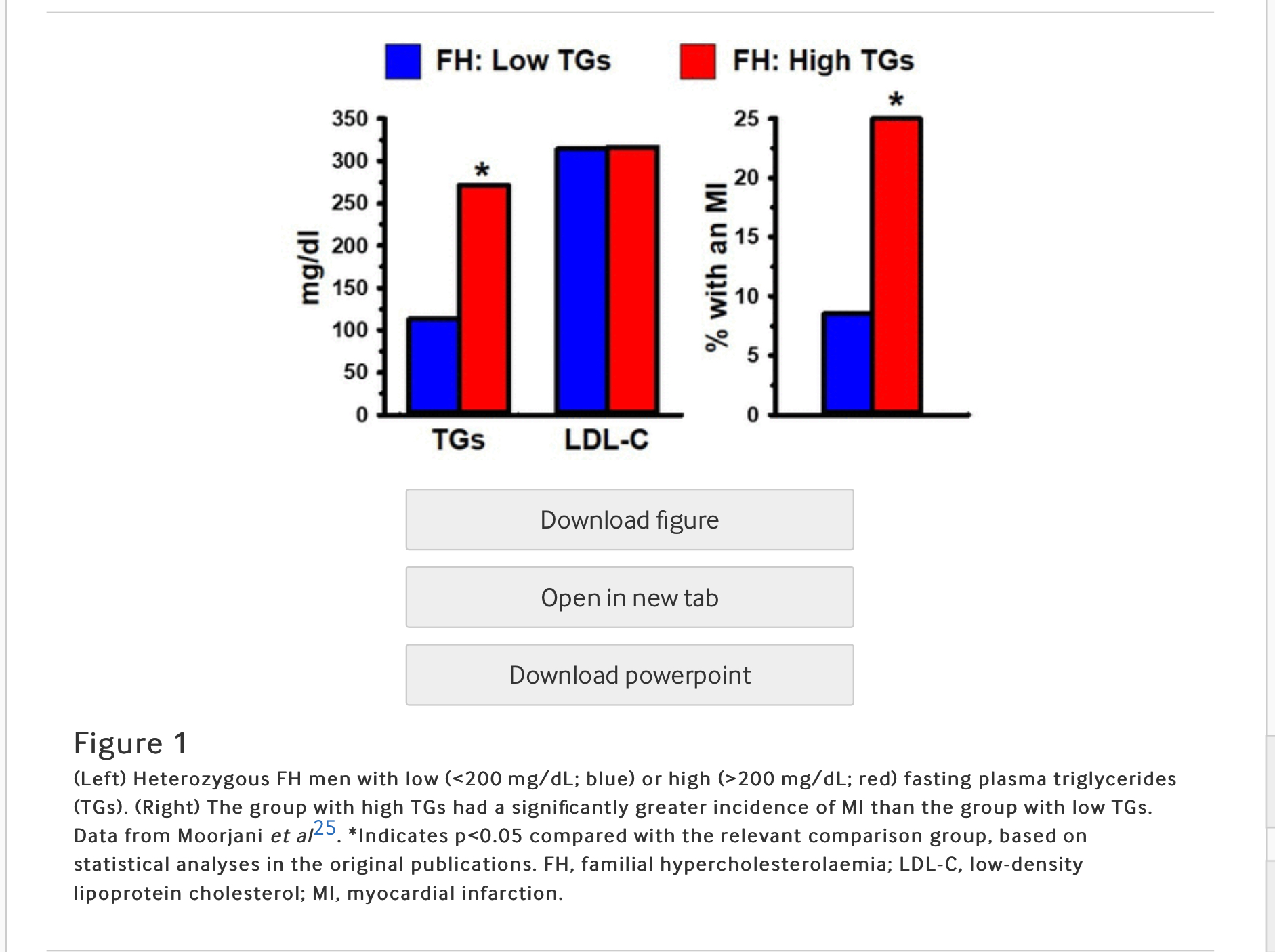
LDL-C is contained in heterogeneous particles which range in size and composition from a small, dense, triglyceride (TG) rich LDL (sdLDL) to a large, buoyant, cholesterol-enriched LDL (lbLDL). This distinction between LDL particle subclasses is important because sdLDL, unlike lbLDL, is a component of an atherogenic dyslipidaemia risk triad (ADRT), composed of elevated levels of TGs and sdLDL, in conjunction with low levels of HDL.8–10 23 Each of the three components of the ADRT, individually, has been associated with increased risk of CHD. For example, sdLDL, unlike lbLDL, is a unique marker of CHD risk, independent of LDL-C.24 Another study demonstrated that FH individuals, distinguished solely on the basis of having high TGs (>200 mg/dL), exhibited three times greater occurrence of a myocardial infarction (MI), compared with FH individuals with low TGs (<200 mg/dL).25 It is noteworthy that the association of high levels of TGs in FH with a high rate of MI occurrence was independent of their LDL-C levels (figure 1). Overall, the ADRT is a highly reliable measure of CHD risk in FH, as well as non-FH, individuals.
Lipoprotein a
Lipoprotein a [Lp(a)] is one of the most robust of all markers of CHD risk in FH and non-FH populations.26 Lp(a) contains a plasminogen-like glycoprotein, known as apolipoprotein (a), which is bound to the apolipoprotein B-100 of an LDL particle. Elevated levels of Lp(a) are more closely associated with CHD than is LDL-C. For example, Seed et al,27 showed that FH individuals with CHD had significantly greater levels of Lp(a) compared with FH without CHD; the association of Lp(a) with CHD in FH was independent of their LDL-C levels (figure 2).
Haemostatic balance between coagulation and fibrinolysis
A powerful influence on the development of CHD is the interplay between processes that promote clot formation (coagulation) and those that cause clots to lyse (fibrinolysis). There is extensive evidence, at cellular, metabolic and genetic levels of analysis, that the haemostatic balance in FH is shifted toward hypercoagulation. These findings were reviewed by Ravnskov et al,18 who found strong evidence of hypercoagulation, and not LDL-C, as a cause of CHD in FH. A subset of the literature is provided below.
Non-lipid CHD risk factors
FH individuals are as susceptible to non-lipid CHD risk factors as non-FH individuals. The following is a subset of the literature that has documented this finding:
Atherogenic dyslipidaemia risk triad: triglycerides, high-density lipoprotein (HDL) and small, dense LDL.
LDL-C is contained in heterogeneous particles which range in size and composition from a small, dense, triglyceride (TG) rich LDL (sdLDL) to a large, buoyant, cholesterol-enriched LDL (lbLDL). This distinction between LDL particle subclasses is important because sdLDL, unlike lbLDL, is a component of an atherogenic dyslipidaemia risk triad (ADRT), composed of elevated levels of TGs and sdLDL, in conjunction with low levels of HDL.8–10 23 Each of the three components of the ADRT, individually, has been associated with increased risk of CHD. For example, sdLDL, unlike lbLDL, is a unique marker of CHD risk, independent of LDL-C.24 Another study demonstrated that FH individuals, distinguished solely on the basis of having high TGs (>200 mg/dL), exhibited three times greater occurrence of a myocardial infarction (MI), compared with FH individuals with low TGs (<200 mg/dL).25 It is noteworthy that the association of high levels of TGs in FH with a high rate of MI occurrence was independent of their LDL-C levels (figure 1). Overall, the ADRT is a highly reliable measure of CHD risk in FH, as well as non-FH, individuals.
Lipoprotein a
Lipoprotein a [Lp(a)] is one of the most robust of all markers of CHD risk in FH and non-FH populations.26 Lp(a) contains a plasminogen-like glycoprotein, known as apolipoprotein (a), which is bound to the apolipoprotein B-100 of an LDL particle. Elevated levels of Lp(a) are more closely associated with CHD than is LDL-C. For example, Seed et al,27 showed that FH individuals with CHD had significantly greater levels of Lp(a) compared with FH without CHD; the association of Lp(a) with CHD in FH was independent of their LDL-C levels (figure 2).
Haemostatic balance between coagulation and fibrinolysis
A powerful influence on the development of CHD is the interplay between processes that promote clot formation (coagulation) and those that cause clots to lyse (fibrinolysis). There is extensive evidence, at cellular, metabolic and genetic levels of analysis, that the haemostatic balance in FH is shifted toward hypercoagulation. These findings were reviewed by Ravnskov et al,18 who found strong evidence of hypercoagulation, and not LDL-C, as a cause of CHD in FH. A subset of the literature is provided below.
- Platelets from FH individuals are more sensitive than those of non-FH individuals to aggregate in response to epinephrine.28 29 This finding suggests that FH individuals would develop a greater thrombotic reaction to stress than non-FH individuals.
- Extremely high levels of fibrinogen, a primary clotting factor and risk factor for CHD,30 are found in homozygous FH individuals, which have a high incidence of early CHD-related mortality.31 High levels of fibrinogen also distinguish the subset of heterozygous FH individuals (as well as non-FH) with CHD from those without CHD (figure 3).30 32
- Genetic factors can influence haemostatic balance. For example, prothrombotic gene polymorphisms, such as prothrombin 20 210A, increase the risk of MI in the general population.33 FH individuals with the prothrombin 20 210A polymorphism exhibited more than twice the rate of coronary events as FH individuals without the polymorphism, an effect which was independent of their LDL levels.34
- FH smokers exhibit a shift in haemostatic balance toward thrombosis, compared with FH non-smokers. Antoniades et al,35 demonstrated that FH smokers exhibited a decreased forearm vasodilatory response to reactive hyperaemia, increased inflammation and an imbalanced thrombosis/fibrinolysis equilibrium favouring hypercoagulation, compared with FH non-smokers.
- Sebestjen et al,36 investigated biomarkers of hypofibrinolysis in FH individuals with and without CHD. They found significantly higher levels of tissue plasminogen activator (PA) antigen and PA inhibitor-1 antigen (both of which suppress fibrinolysis) in FH individuals with CHD. This shift of haemostatic balance from fibrinolysis toward hypercoagulation was independent of their LDL levels.
Non-lipid CHD risk factors
FH individuals are as susceptible to non-lipid CHD risk factors as non-FH individuals. The following is a subset of the literature that has documented this finding:
- Galema-Boers et al37 demonstrated that FH individuals with hypertension had more than twice the incidence of CHD than normotensive FH individuals, despite having equivalent LDL-C levels.
- Miname et al38 found that FH individuals with a high CAC score, which is a highly reliable marker of CHD, had significantly greater levels of fasting blood glucose than those with low CAC, but both groups had equivalent on-treatment levels of LDL-C.
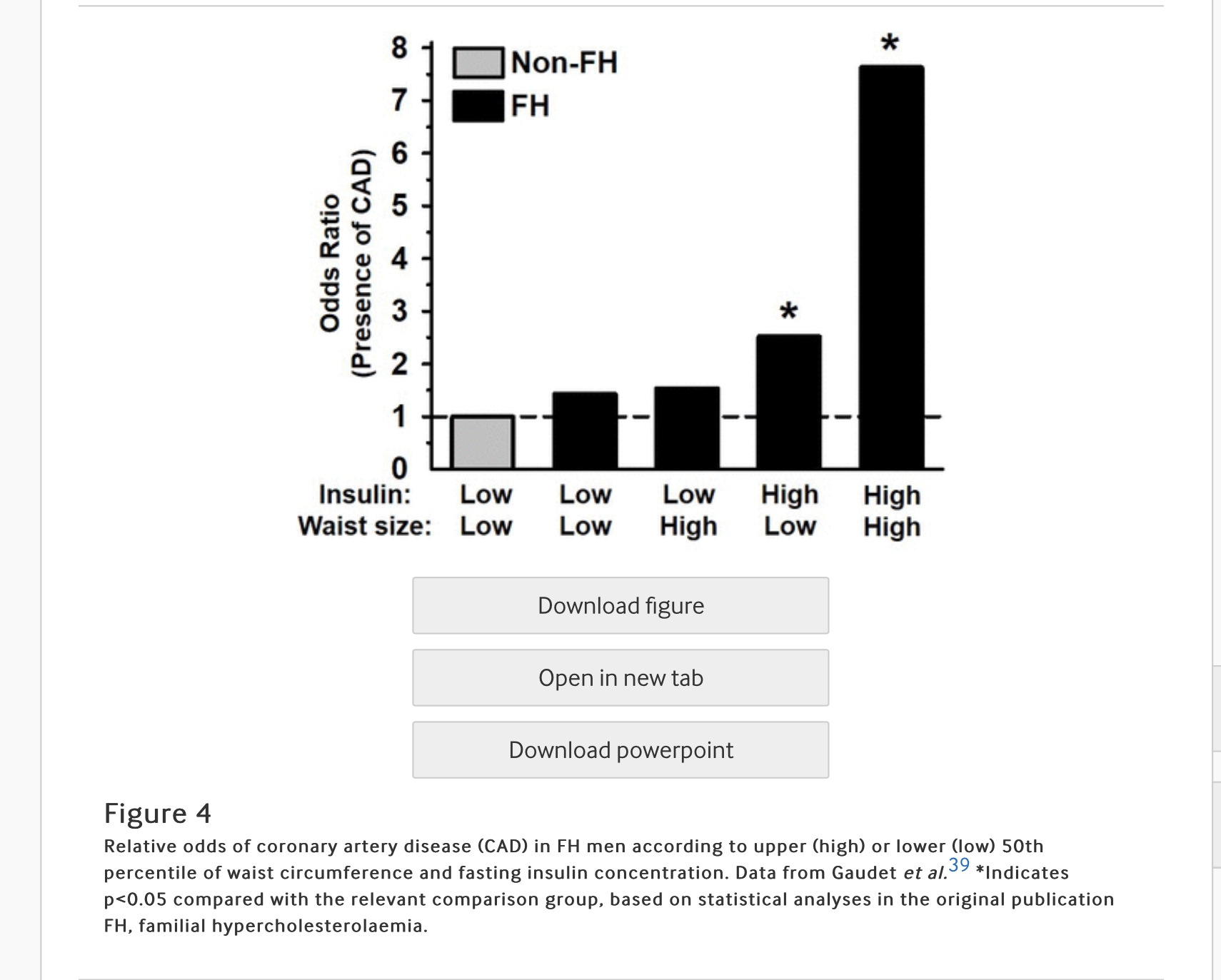
- Gaudet et al39 reported that FH individuals with abdominal obesity and hyperinsulinemia exhibited a dramatically greater incidence of coronary artery disease (CAD) than FH individuals without abdominal obesity and hyperinsulinaemia, an effect which was independent of their LDL levels (figure 4).
- Ye et al40 found that patients with FH with detectable CAC had significantly increased high‐sensitivity C reactive protein (hsCRP) values and impaired flow-mediated dilation compared with FH patients without CAC, which was independent of LDL-C levels.
- A subset of FH individuals display elevated TGs, in addition to increased LDL-C, a condition referred to as FH type IIb25 or familial combined hyperlipidaemia (FCH).41 The susceptibility of FCH individuals to develop CHD is influenced by genetics and obesity.42 43 FCH individuals exhibit a significantly higher rate of MI than FH individuals with low levels of TGs, despite having equivalent levels of LDL-C.25 Therefore, hypertriglyceridaemia and obesity, independent of LDL-C, in a subset of FH individuals increases their susceptibility to develop CHD.
- Click here for summary of this paper.
 RSS 訂閱
RSS 訂閱

{kind=link}
{kind=link}
{kind=link}